29 August 2022: Database Analysis
Transcriptome Analysis of Peripheral Blood Mononuclear Cells Response in Patients with Severe COVID-19 Reveals Crucial Genes Regulating Protein Ubiquitination
Jinfeng Huang1ABCDEF, Ruobing Li2EF, Jie Cheng3B*, Peilin Zhen1ABDOI: 10.12659/MSM.937532
Med Sci Monit 2022; 28:e937532






Abstract
BACKGROUND: We sought to further our understanding of the biological characteristics underlying severe COVID-19.
MATERIAL AND METHODS: RNA sequencing (RNA-Seq) analysis was used to evaluate peripheral blood mononuclear cells from 4 patients with severe COVID-19 and 4 healthy controls. We performed gene expression analyses to detect differentially expressed genes (DEGs). Enrichment analyses were performed to identify their molecular processes and signaling pathways, and the protein–protein interaction network was constructed to extract the core gene cluster. The investigation of protein-chemical interactions and regulatory signatures for specific regulatory checkpoints and powerful chemical agents was then conducted for these essential genes. Finally, we used single-cell RNA-Seq analysis from an online platform to show how these genes were expressed differently, depending on the kind of cell.
RESULTS: A total of 268 DEGs were found. The biological process of protein ubiquitination was later discovered to be highly influenced by the core gene cluster (ITCH, TRIM21, RNF130, FBXO11, UBE2J1, and ASB16) at the transcriptome level. Six transcription factors, FNIC, FOXA1, YY1, GATA2, MET2A, and FOXC1, as well as miRNAs hsa-miR-1-3p and hsa-miR-27a-3p were identified. We found a potent chemical agent, copper sulfate, may regulate protein ubiquitination genes cooperatively, and the genes regulating protein ubiquitination could be expressed highly on the macrophages.
CONCLUSIONS: Taken together, we suggest that protein ubiquitination is a crucial functional process in patients with severe COVID-19. This study will give a deeper insight into biological characteristics and progression of COVID-19 and facilitate development of novel therapeutics, leading to significant advancements in the COVID-19 pandemic.
Keywords: COVID-19, Transcriptome, ubiquitination, COVID-19, F-Box Proteins, Gene Expression Profiling, Gene Regulatory Networks, Humans, Leukocytes, Mononuclear, MicroRNAs, Pandemics, Protein-Arginine N-Methyltransferases
Background
COVID-19 is a respiratory condition caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which belongs to the Coronaviridae family and whose genome is composed of a single 30-kb strand of RNA [1]. As of August 2, 2022, there have been 583 238 204 confirmed COVID-19 cases, and it is reported that there have been 6 422 235 COVID-19-related deaths across 223 nations or regions (https://worldometers.info/coronavirus/). The symptoms mainly manifest as inflammatory lesions in the lungs. The virus also damages the digestive system, nervous system, and cardiovascular systems of patients, and an infection could result in death due to multiple organ failure [2]. It was reported that 80% of patients were asymptomatic or had mild disease, while about 14% had severe disease, and 5% were in critical condition [3]. The grade of severity in COVID-19 was related to the age, sex, and other health conditions of the patient, such as the occurrence of chronic diseases, including diabetes and heart disease [4]. Therefore, it is particularly important to obtain knowledge regarding the treatment of patients with severe and critical disease, as such patients are more likely to deteriorate rapidly, resulting in cytokine (inflammatory) storms, respiratory distress, multi-organ dysfunction, and potential death [2].
Mutations always occur during the spread of viruses. Mutations were especially observed to occur in patients infected with the delta variant, which is highly infectious and has resulted in more severe illness in individuals after it originated in India in December 2020 [5]. Vaccination is reported to be the most effective method for the prevention of COVID-19 disease [6], since no dedicated antiviral drugs are available for the treatment of patients with severe COVID-19, and the use of drugs is limited to the management of symptoms and complications. Therefore, it is important to obtain insights into the characteristics and molecular mechanisms of biological reactions occurring in patients with COVID-19 in order to develop new and efficacious clinical therapeutic targets.
The SARS-CoV-2 genome contains 4 major structural proteins, namely the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins [7]. The S1 subunit region of the S protein facilitates the binding of the virus to the angiotensin-converting enzyme 2 receptor on the surface of human lung cells [8]. After the virus enters the cells, an immune response is generated by the host, and pattern recognition receptors, such as toll-like receptors that are present on antigen-presenting cells (APCs), receive signals [9].
Post-translational modifications are chemical modifications necessary for precise regulation of protein properties and functions, which greatly increase the complexity and functional diversity of the proteome and ensure the dynamic cellular response to the external environment and intracellular factors [10]. Ubiquitination is a post-translational modification that is achieved through a coordinated and sequential enzymatic cascade. Ubiquitin is a 76-amino acid protein that is activated by the ubiquitin-activating (E1) enzyme in an ATP-dependent reaction. Ubiquitin is subsequently transferred to the active Cys residue of the ubiquitin-binding (E2) enzyme, and is then attached to the substrate mediated by the ubiquitin-binding (E3) enzyme. So far, 2 E1s, nearly 30 E2s, and more than 600 E3s have been found in humans [11]. Protein ubiquitination is an essential process in several eukaryotic biological processes, including cellular proteostasis, DNA repair, trafficking, immune functions, and antiviral responses [12,13]. Ubiquitination plays a key role in viral replication and affects the virulence of the influenza virus via the production of infectious virus particles in the cytoplasmic domain of influenza A virus membrane protein 2 [14]. It was reported that the ubiquitination of Beclin1 would inhibit autophagy and reduce MERS-coronavirus infections [15]. Ubiquitination has also has been shown to affect the pathogenesis of acute lung injury and other lung diseases and to particularly affect the functioning of the alveolar epithelial barrier [16]. Siu et al demonstrated that SARS-CoV open reading frame 3a (ORF3a) mediated the functioning of the NLRP3 inflammasome via TRAF3-dependent ubiquitination [17]. Furthermore, SARS-CoV-2 ORF7a usurps the ubiquitination system of the host and promotes the antagonism attributable to the interferon I response [18].
Itchy E3 ubiquitin protein ligase (ITCH) could play a role in innate immunity. The ITCH substrates would act as central participants for accelerating the degradation of c-Jun and JunB [19], and ubiquitinating PLC-γ1 and PKC-θ for T cell anergy [20]. In addition, ITCH might enhance the termination of antiviral responses via ubiquitination and degradation of MAVS [21]. Tripartite motif protein (TRIM21) is an E3 ligase that enhances target specificity during ubiquitination. In the pro-inflammatory environment, it would translocate to the nucleus rather than to the cytoplasmic environment [22]. Das et al suggested that TRIM21 interacts with the complex of IFI35 and Nmi in an uncontrolled anti-viral signaling environment [23]. Ring finger proteins (RNF) account a large class of potential ubiquitin ligases, and it is shown that RNF130 could negatively regulate T-cell receptor signaling in T cells infected with the human T-cell lymphotropic virus-1 [24]. Christof et al suggested that the inactivation of F-box only protein 11 (FBXO11) as a tumor suppressor was related to the occurrence of lymphoproliferative pathologies [25]. It might be crucial for the release of PRC2-mediated repression throughout the maturity of the erythroid [25]. Ubiquitin-conjugating enzyme E2 J1 (UBE2J1) is an E2 (ubiquitin-conjugating) enzyme involved in processes involving endoplasmic reticulum-associated protein degradation on the endoplasmic reticulum, where it could degrade incorrectly folded proteins. UBE2J1 could regulate the interferon pathway and degrade the promotion of infection with RNA viruses [26]. Most reports on ASB16 have primarily focused on lncRNA ASB16 antisense RNA 1, as it is recognized as an oncogene in several types of cancer, including gastric cancer [27], hepatocellular carcinoma, and glioma [28,29]. For instance, in gastric cancer, ASB16-AS1 would regulate the TRIM37 levels to induce the NF-κB pathway and promote tumor proliferation [27].
Copper has a powerful ability to neutralize infectious viruses, such as bronchitis virus, poliovirus, human immunodeficiency virus type 1 (HIV-1), and other enveloped or non-enveloped single- or double-stranded DNA and RNA viruses [30,31] because copper can damage the genomic DNA and RNA of the virus [32]. Moreover, Doremalen et al indicated that SARS-CoV-2 is very sensitive to copper [33]. In clinical practice, copper sulfate is frequently used locally as a fungicide, bactericide, and astringent [34].
In this study, we pre-processed raw data from the Gene Expression Omnibus (GEO) GSE167930 dataset and identified differentially expressed genes (DEGs) to investigate biological features and progression in patients with severe COVID-19. The functions and signaling pathways of these DEGs were identified to gain knowledge regarding their molecular processes. Then, we examined core gene clusters in patients with severe COVID-19 by performing protein–protein interaction (PPI) network analysis and determined the most significant gene cluster. Genes in this cluster were found to be involved in the biological process of protein ubiquitination. Furthermore, using these identified genes at regulatory checkpoints, we recognized microRNAs and transcription factors. Moreover, to specify the potent chemical agents responsible for protein ubiquitination in patients with severe COVID-19, we conducted an analysis of the protein-chemical interaction network. Finally, using data from a public dataset, we performed single-cell RNA sequencing (RNA-seq) analysis to show how these genes are expressed differently in different cell types. Figure 1 depicts the sequential workflow of this study.
Material and Methods
DATASETS, DATA PRE-PROCESSING, AND DIFFERENTIAL EXPRESSION ANALYSIS:
We used RNA-Seq datasets of 4 patients with severe COVID-19 and 4 healthy controls, from whom peripheral blood samples were gathered, from the GEO database of the National Center for Biotechnology Information. The GEO accession number of the dataset was GSE167930, and it was already deposited for the earlier study our team published [35]. The patient information is provided in Table 1. To identify significant DEGs, the edgeR package by online OmicShare tools was used to analyze the RNA-Seq count dataset. The edgeR package operates on a read count table where the columns represent independent libraries and the rows represent genes. The counts show how many reads have been aligned to each gene overall. After the normalization process, the significant DEGs associated with severe COVID-19 were filtered if P<0.05 and |log2 FC| ≥1. The OmicShare tool, a free online platform for data analysis (https://www.omicshare.com/tools), was used to perform the differential expression analysis and create the graphics. The volcano plot was plotted with log2(FC) as the horizontal coordinate and −log10(P value) as the vertical coordinate, and points with significant differences were given different colors to show the significance of the difference between the severe COVID-19 and healthy control groups. The heatmap aggregated the gene expression abundance of 4 patients with severe COVID-19 and 4 healthy controls. After Z-score normalization of the rows, the progressive color was used to visualize the gene expression abundance and thus the pattern of variation of different genes in the samples.
GENE ONTOLOGY AND KYOTO ENCYCLOPEDIA OF GENES AND GENOMES ENRICHMENT ANALYSIS:
To characterize biological mechanisms, gene ontology (GO) and functional enrichment (biological processes, cellular component, and molecular functions) analyses were performed, to analyze the DEGs in patients with severe COVID-19 using the database for annotation, visualization, and integrated discovery (DAVID) v6.8 (
GENE SET ENRICHMENT ANALYSIS:
Gene set enrichment analysis (GSEA) was used to analyze the genomic information of each sample according to its degree of differential expression. GSEA identified whether the preset gene set was enriched at the top or bottom of the sequencing table by sequencing the genes by the degree of differential expression between the severe COVID-19 and healthy control groups. Since the analysis examines genome expression rather than the expression of specific genes, it can take into account more subtle expression changes. To obtain insight into the signaling and metabolic molecules involved in biological pathways, all the genetic information regarding patients with severe COVID-19 and healthy controls was uploaded onto GSEA software, and analysis was performed using the Reactome pathway database. A standard metric was applied based on a normalized enrichment score (NES) >1,
PPI NETWORK ANALYSIS AND GENE CLUSTER IDENTIFICATION:
To construct the PPI network interaction diagram, the DEGs identified in severe COVID-19 samples were uploaded to STRING. Then, the results obtained using STRING were uploaded into Cytoscape v.3.7.1., and the molecular complex detection (MCODE) plug-in of Cytoscape was used for cluster analysis. Next, the genes in the cluster with the highest scores were imported into DAVID to determine the biological processes in which this gene cluster participated significantly. Subsequently, genes identified to be involved in the most significant biological process were imported into the NetworkAnalyst tool for further analysis.
REGULATORY SIGNATURES NETWORK ANALYSIS:
To detect the transcriptional signatures and post-transcriptional regulatory signatures of genes involved in protein ubiquitination, the interacting network of transcription factors and miRNAs were identified using the NetworkAnalyst tool. The JASPAR database was used for transcription factors, and the Tarbase database was used for miRNAs.
PROTEIN–CHEMICAL INTERACTION NETWORK ANALYSIS:
To identify potential antiviral agents, the protein-chemical interaction network was identified using the Comparative Toxicogenomics Database in the NetworkAnalyst tool. Cytoscape was used to visualize this interaction network.
SINGLE-CELL RNA-SEQ ANALYSIS:
We used single-cell RNA-Seq analysis on a free online database platform named “Large-scale single-cell analysis reveals critical immune characteristics of COVID-19 patients” (
Results
SAMPLE INFORMATION PROCESSING AND SCREENING OF DEGS:
To identify the characteristics of severe COVID-19 in this study, we analyzed the RNA-Seq count dataset using the edgeR package and filtered the DEGs if the P value was <0.05 and |log2 FC| ≥1; we obtained a count dataset of 10 894 genes and 268 DEGs (Figure 2A). The gene cluster in each patient with severe COVID-19 and healthy controls is shown on the heatmap (Figure 2B).
ENRICHMENT ANALYSIS:
The GO project is a resource with extensive computable knowledge that focuses on the functions of genes and their products. Here, we performed GO enrichment analysis for the 268 DEGs identified in patients with severe COVID-19 with regard to 3 categories: biological process, cellular component, and molecular function. Figure 3 shows the top 10 terms in each category. For molecular function (Figure 3A), the most significant term was “amide transmembrane transporter activity”; and the “intracellular part” was mostly enriched in the cellular component (Figure 3B). Then, the “isopentenyl diphosphate biosynthetic process” and “mevalonate pathway” played a significant role in the biological process (Figure 3C). To demonstrate the biochemical processes in patients with severe COVID-19, pathway analysis was performed using the KEGG and Reactome databases. As shown in Figure 3D, there were 8 significant pathways in KEGG, including “terpenoid backbone biosynthesis”, “fanconi anemia pathway”, and “endocytosis”. Figure 4 shows the top 3 pathways in the Reactome database, which were identified using GESA software. We observed that 17, 21, and 19 genes were enriched significantly in “insulin receptor recycling” (Figure 4A), “latent infection of homo sapiens with Mycobacterium tuberculosis” (Figure 4B), and “transferrin endocytosis and recycling” (Figure 4C).
CONSTRUCTION OF THE PPI NETWORK AND FURTHER EXCAVATION OF GENE CLUSTERS INVOLVED IN THE PROTEIN UBIQUITINATION BIOLOGICAL PROCESS:
To acquire insights into the network of reactions attributable to DEGs in severe COVID-19 samples, 268 DEGs were uploaded to the STRING database. We observed that 261 nodes and 307 edges were obtained with PPI enrichment at a P value of 0.01. Figure 5 shows this data file, which was obtained using Cytoscape. To further identify the core genes playing key roles, MCODE was used to obtain the network data. Table 2 presents the 9 identified gene clusters. The genes in the cluster with the highest score (ANAPC1, TRIM21, ASB16, RNF130, FBXO11, UBE2J1, FBXO41, and ITCH) were further selected for biological process enrichment analysis to determine which pathway was most involved in the process. The highest statistical significance was observed for the biological process “protein ubiquitination” with 6 genes, ITCH, TRIM21, RNF130, FBXO11, UBE2J1, and ASB16, based on the P values and FDR (Table 3). These 6 critical genes involved in protein ubiquitination were analyzed further.
REGULATORY SIGNATURES AND PROTEIN-CHEMICAL INTERACTIONS BETWEEN GENES INVOLVED IN PROTEIN UBIQUITINATION:
To identify the more substantial mechanisms at the transcriptional level of genes involved in the protein ubiquitination pathway during severe COVID-19, networks of regulatory signatures (transcription factors and miRNAs) were constructed. Figure 6 shows that a total of 26 transcription factors might influence genes ASB16, UBE2J1, TRIM21, ITCH, and FBXO11 and regulate protein ubiquitination during severe COVID-19. Six transcription factors, FNIC, FOXA1, YY1, GATA2, MET2A, and FOXC1, linked more than 2 genes. Among these miRNAs, hsa-mir-1-3p and hsa-mir-27a-3p were also found to significantly exert regulation on over 5 of the genes involved in protein ubiquitination. The miRNA hsa-mir-1-3p was connected with ITCH, TRIM21, RNF130, FBXO11, and UBE2J1, and hsa-mir-27a-3p was associated with ITCH, RNF130, FBXO11, UBE2J1, and ASB16 (Figure 7). We also identified 64 chemical agents related to ubiquitination that could be used for identifying potential antiviral agents; among them, 21 chemical agents were associated with more than 2 genes. Copper sulfate was found to significantly correlate with 5 genes, ITCH, TRIM21, RNF130, FBXO11, and ASB16 (Figure 8).
SINGLE-CELL RNA-SEQ ANALYSIS:
The expression of the 6 genes (ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16) regulating protein ubiquitination in patients with severe COVID-19 was evaluated using an online single-cell RNA-Seq platform. Eight cell types were identified, including CD8 T cells, CD4 T cells, B cells, monocyte, natural killer cells, macrophages, neutrophils, and dendritic cells (Figure 9A). Figure 9B shows the integration of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells, and they were obviously enriched most significantly in macrophages, with a mean of 0.306. Among these genes, as shown in Figure 9C, ITCH was mostly enriched in macrophages, with a mean of 0.234; TRIM21 was expressed highly in macrophages, with a mean of 0.157; RNF130 was also mostly enriched in macrophages, with a mean of 0.874; UBE2J1 was also highly expressed in macrophages, with a mean of 0.547;FBXO11 was highly enriched in macrophages, with a mean of 0.092, while ASB16 was enriched rarely in all cell types and it was expressed mostly in CD8 T cells, with a mean of 0.003. Subsequently, Figure 9D shows the scatter plots of all the cells with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression, and Figure 9E shows the scatter plots of macrophages. The above results indicated that the genes regulating protein ubiquitination in patients with severe COVID-19 could be expressed on the macrophages.
Discussion
SARS-CoV-2 has continued to remain a threat worldwide. Almost 10% to 20% of patients infected with SARS-CoV-2 progress to severe COVID-19 with pneumonia, respiratory failure, and potentially fatal situations, although most individuals infected with SARS-CoV-2 are usually asymptomatic or have mild symptoms in Wuhan, China [36]. Hence, it is important to understand the characteristics and molecular mechanisms of the biological reactions occurring in patients with severe COVID-19 thoroughly, for the development of biomarkers and therapeutic targets. In this study, we identified a network-based framework that helped us gain insight into the gene expression patterns in peripheral blood samples obtained from patients with severe COVID-19. RNA-Seq is considered significantly important in biomedical research. We used this method to identify 268 significant DEGs in patients with severe COVID-19. To characterize molecular processes, these DEGs were uploaded into the GO, KEGG, and Reactome databases to identify gene functions and signaling pathways, and PPI network analysis was performed. To focus on the core gene clusters, we detected 6 genes (
Patients with severe COVID-19 were more likely to have underlying comorbidities, such as diabetes (7.3%), cancer (5.6%), hypertension (6.0%), and a higher fatality rate, according to the American College of Cardiology clinical bulletin [2,37] ]. Yang et al revealed that the most distinctive comorbidities of the critically ill adult pneumonia patients with SARS-CoV-2, who were admitted to the intensive care unit, were cerebrovascular diseases (22%) and diabetes (22%) [38]. Interestingly, in our study, we identified 17 genes that were significantly enriched in the insulin receptor recycling pathway that played a key role in regulating diabetes. This could be considered as evidence indicating the relationship between diabetes and severe COVID-19 and might be related to insulin receptor recycling. It has been reported that infection with
The MCODE plugin of Cytoscape is widely used for finding highly interconnected regions in a PPI network based on topology. Using this algorithm, 1 cluster, including 8 genes (
It was reported that the phosphorylation of the
Furthermore, to obtain more information regarding the mechanisms for ubiquitination development in patients progressing to severe COVID-19, transcription factors and miRNAs were detected at the post-transcription level. In this regard, we identified 26 transcription factors, including FNIC, FOXA1, YY1, GATA2, MET2A, and FOXC1 transcripts, from more than 2 genes. Surprisingly, this result was in accordance with those observed after the regulatory network analysis of lung epithelial cells in COVID-19 by Islam et al, in which significantly important transcription factors, such as FOXC1, GATA2, YY1, FOXL1, and NFKB1, were identified [48].
Conclusions
In the present study, we aimed to provide insight into the potential pathogenic processes in patients with severe COVID-19 that might facilitate the development of novel and effective clinical treatment targets. We first determined 268 DEGs in patients with severe COVID-19. Using these DEGs, we identified key molecular processes and signaling pathways. Then, based on the PPI network, we further identified the most significant gene cluster, which was found to be involved in protein ubiquitination. The 6 genes
Figures
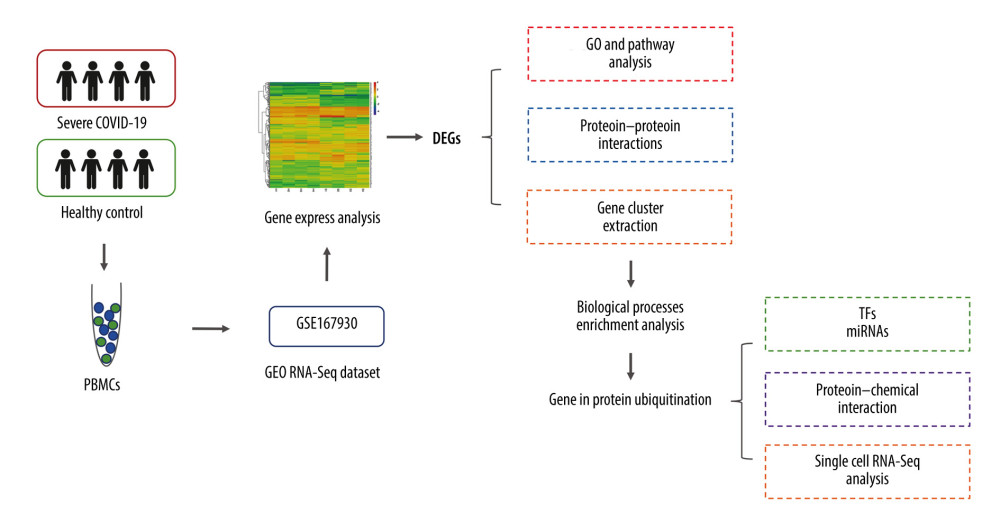 Figure 1. Diagrammatic representation of the overall general process flow of this study showing the RNA sequencing (RNA-Seq) datasets of 4 patients with severe COVID-19 and 4 healthy controls, from whom peripheral blood samples were obtained from GSE167930. Then, the raw data were processed to identify the differentially expressed gene (DEGs). We used these DEGs for Gene Ontology and pathway analysis and protein–protein interaction (PPI) analysis. The most significant gene cluster in the PPI network was identified and used for biological process enrichment analysis. Genes in this cluster were found to play a role in protein ubiquitination. Then, the analysis of genes involved in protein ubiquitination was performed to identify the network of protein-chemical interactions for potent chemical agents, as well as transcription factors and microRNAs at regulatory checkpoints. Ultimately, single-cell RNA-Seq analysis of an online dataset was carried out to reveal the expression of the genes regulating protein ubiquitination that is specific to different cell types.
Figure 1. Diagrammatic representation of the overall general process flow of this study showing the RNA sequencing (RNA-Seq) datasets of 4 patients with severe COVID-19 and 4 healthy controls, from whom peripheral blood samples were obtained from GSE167930. Then, the raw data were processed to identify the differentially expressed gene (DEGs). We used these DEGs for Gene Ontology and pathway analysis and protein–protein interaction (PPI) analysis. The most significant gene cluster in the PPI network was identified and used for biological process enrichment analysis. Genes in this cluster were found to play a role in protein ubiquitination. Then, the analysis of genes involved in protein ubiquitination was performed to identify the network of protein-chemical interactions for potent chemical agents, as well as transcription factors and microRNAs at regulatory checkpoints. Ultimately, single-cell RNA-Seq analysis of an online dataset was carried out to reveal the expression of the genes regulating protein ubiquitination that is specific to different cell types. 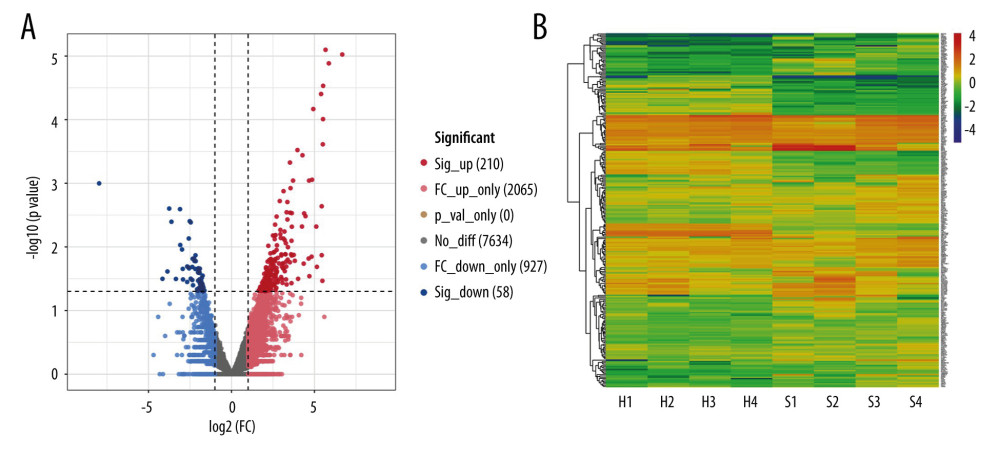 Figure 2. Differentially expressed genes in patients with severe COVID-19 and healthy control samples are shown in the (A) volcano plot and (B) heatmap. H – healthy control; S – severe COVID-19.
Figure 2. Differentially expressed genes in patients with severe COVID-19 and healthy control samples are shown in the (A) volcano plot and (B) heatmap. H – healthy control; S – severe COVID-19. 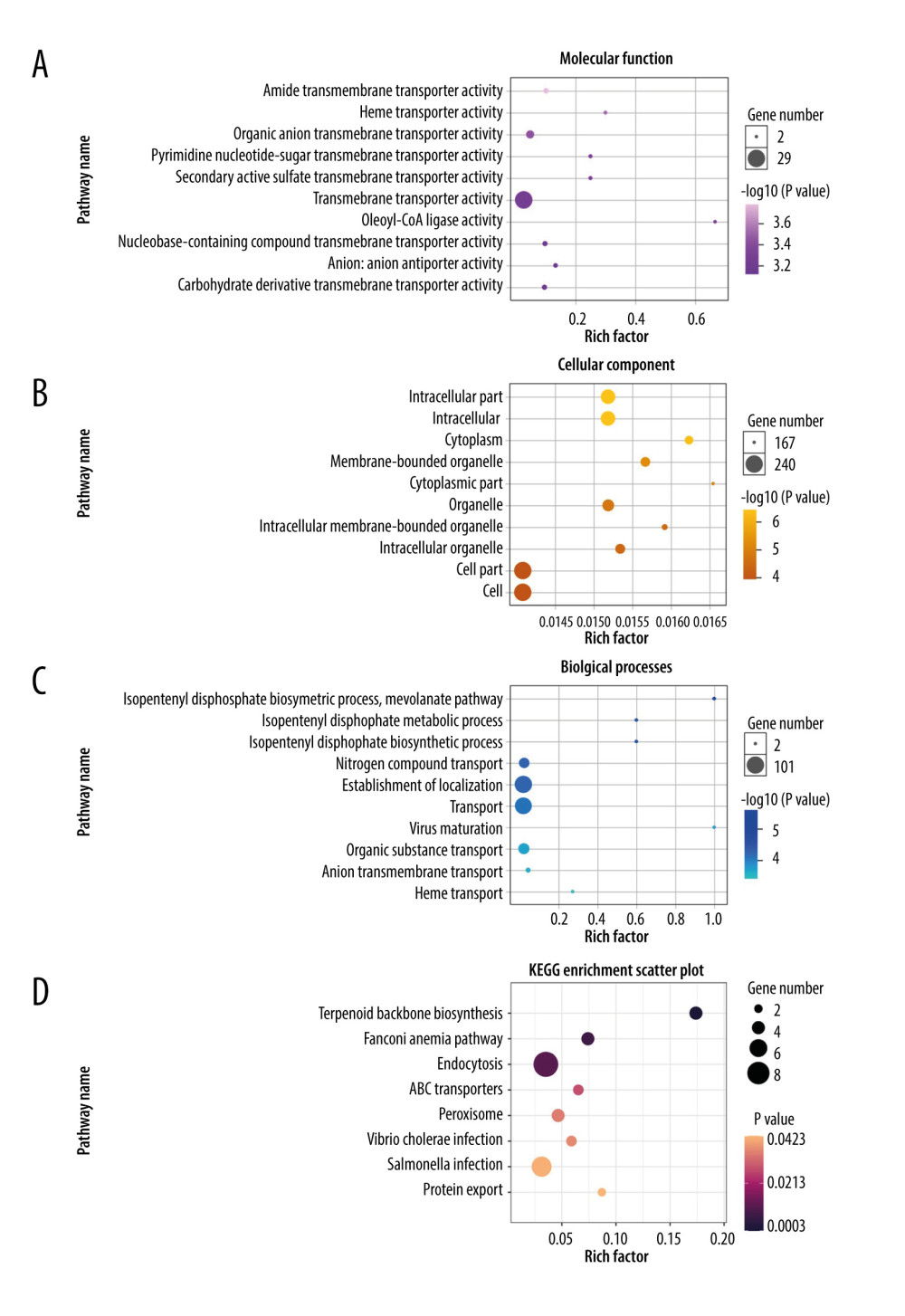 Figure 3. Gene Ontology and KEGG enrichment analysis of 268 differentially expressed genes (DEGs) in severe COVID-19. The top 10 terms in (A) molecular function, (B) cellular component, and (C) biological process category are presented. The total of 8 significant pathways involved in (D) KEGG enrichment are shown using a scatter plot. Colors represent the P values and sizes of the spots represent the counts of genes.
Figure 3. Gene Ontology and KEGG enrichment analysis of 268 differentially expressed genes (DEGs) in severe COVID-19. The top 10 terms in (A) molecular function, (B) cellular component, and (C) biological process category are presented. The total of 8 significant pathways involved in (D) KEGG enrichment are shown using a scatter plot. Colors represent the P values and sizes of the spots represent the counts of genes. 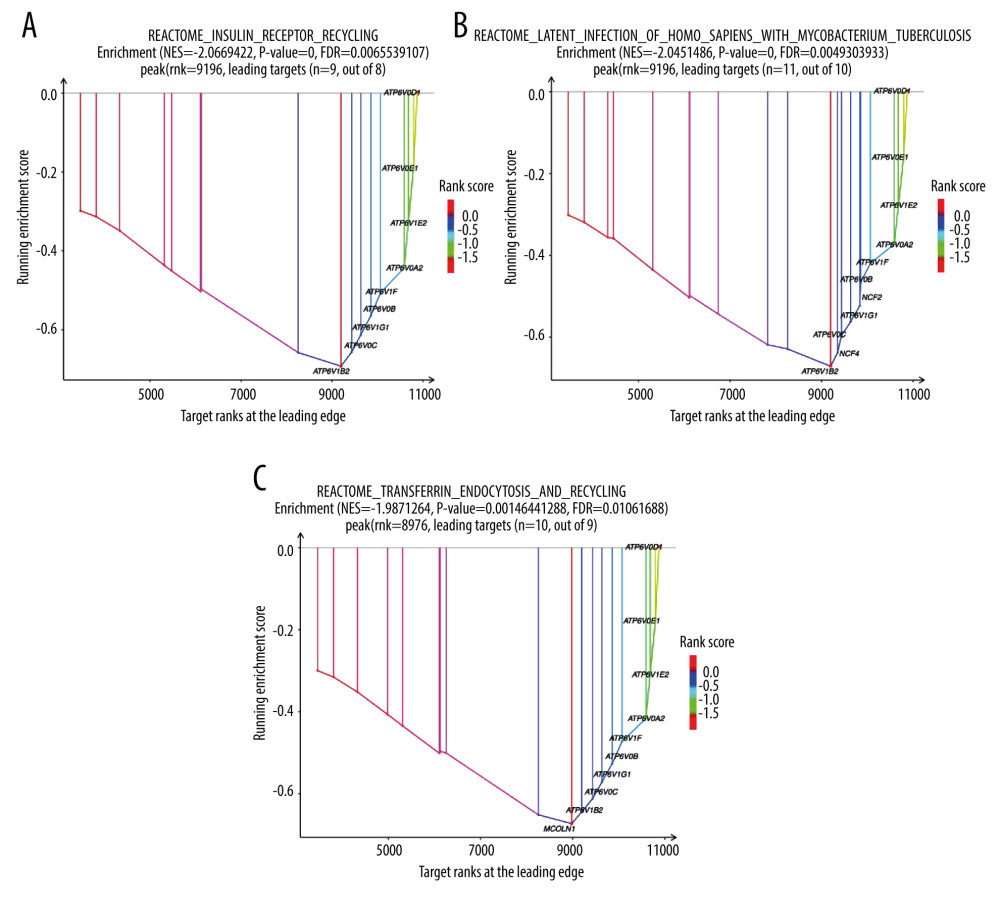 Figure 4. The top 3 pathways in the Reactome database. The gene set was filtered using gene set enrichment analysis, based on the number of genes contained in the gene set, with a minimum number of 15 genes and a maximum number of 500 genes by default. (A) REACTOME_INSULIN_RECEPTOR_RECYCLING, (B) REACTOME_LATENT_INFECTION_OF_HOMO_SAPIENS_WITH MYCOBACTERIUM_TUBERCULOSIS, (C) REACTOME_TRANSFERRIN_ENDOCYTOSIS _AND_RECYCLING.
Figure 4. The top 3 pathways in the Reactome database. The gene set was filtered using gene set enrichment analysis, based on the number of genes contained in the gene set, with a minimum number of 15 genes and a maximum number of 500 genes by default. (A) REACTOME_INSULIN_RECEPTOR_RECYCLING, (B) REACTOME_LATENT_INFECTION_OF_HOMO_SAPIENS_WITH MYCOBACTERIUM_TUBERCULOSIS, (C) REACTOME_TRANSFERRIN_ENDOCYTOSIS _AND_RECYCLING. 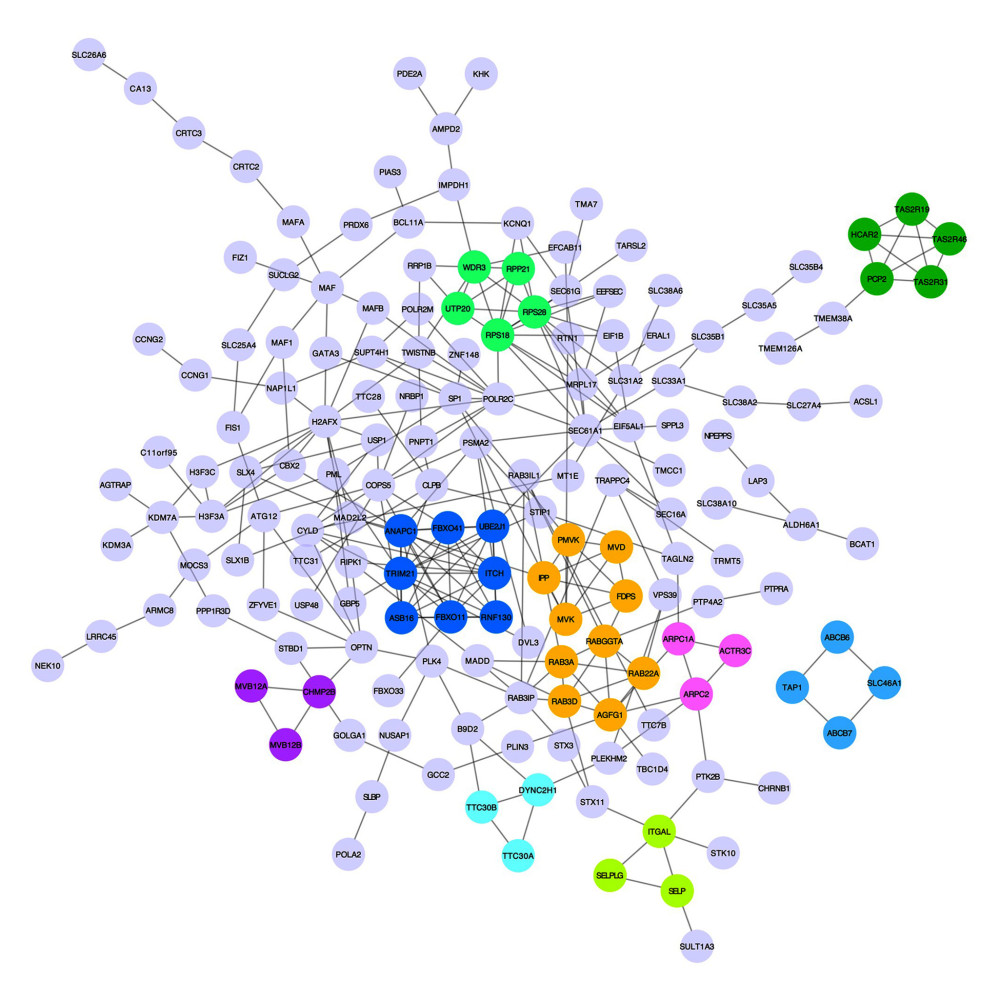 Figure 5. A network of protein–protein interactions (PPI) was built using STRING and Cytoscape. Different colors are used to denote the various clusters that MCODE examined. The PPI network has 261 nodes and 307 edges.
Figure 5. A network of protein–protein interactions (PPI) was built using STRING and Cytoscape. Different colors are used to denote the various clusters that MCODE examined. The PPI network has 261 nodes and 307 edges. 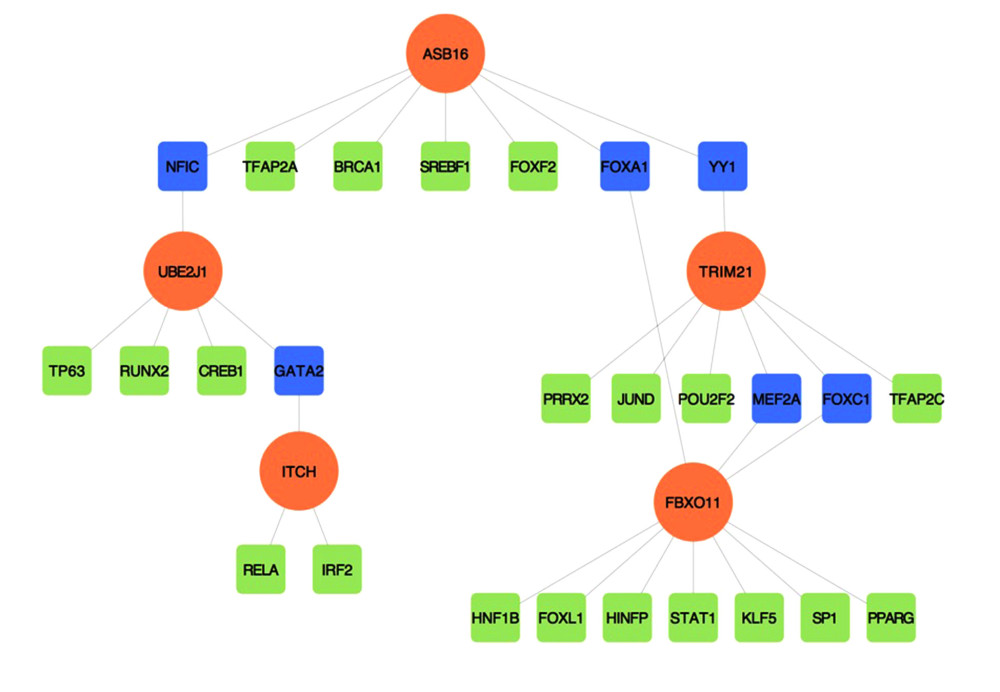 Figure 6. The regulatory interaction network of gene-transcription factors was established with network analyst. Genes are represented by circle nodes and are colored orange; transcription factors are represented by square nodes and are colored green; transcription factors that simultaneously target more than 2 genes are represented by blue nodes.
Figure 6. The regulatory interaction network of gene-transcription factors was established with network analyst. Genes are represented by circle nodes and are colored orange; transcription factors are represented by square nodes and are colored green; transcription factors that simultaneously target more than 2 genes are represented by blue nodes. 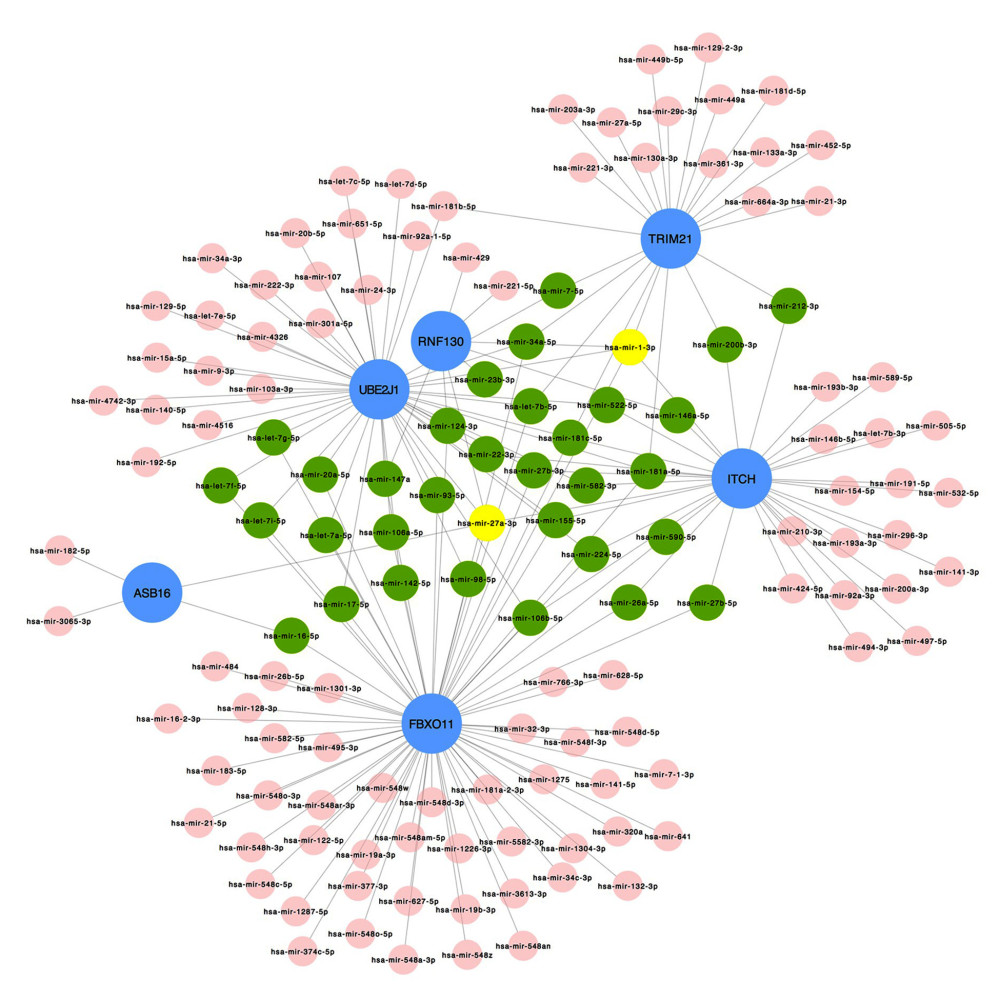 Figure 7. Interaction network between genes involved in protein ubiquitination and targeted miRNAs. Genes are shown in blue; miRNAs are shown in pink; miRNAs targeting more than 2 genes simultaneously are shown in green; miRNAs targeting 5 genes simultaneously are shown in yellow. There are 134 nodes and 187 edges.
Figure 7. Interaction network between genes involved in protein ubiquitination and targeted miRNAs. Genes are shown in blue; miRNAs are shown in pink; miRNAs targeting more than 2 genes simultaneously are shown in green; miRNAs targeting 5 genes simultaneously are shown in yellow. There are 134 nodes and 187 edges. 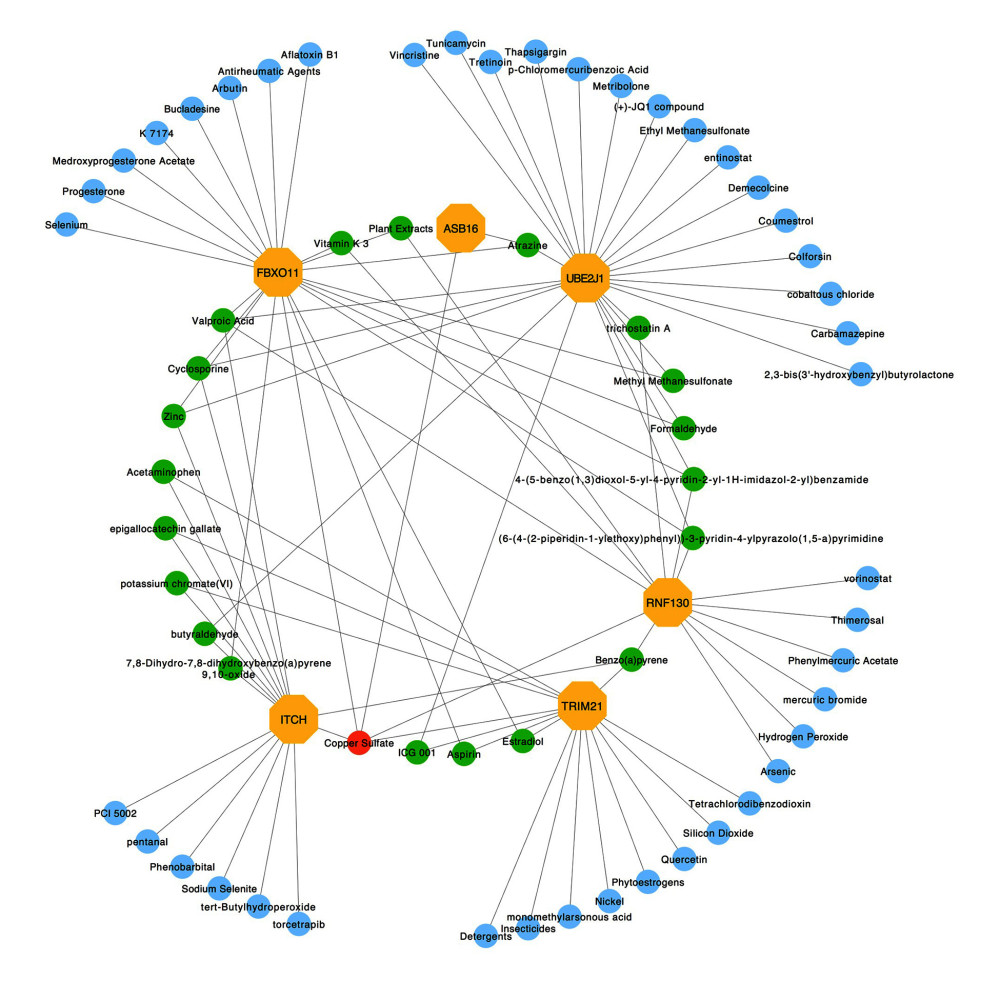 Figure 8. Protein–chemical interaction network. Of a total of 70 nodes, genes are colored in yellow, chemical agents are colored in blue, chemical agents that target more than 2 genes at once are shown in green, and chemical agents that target 5 genes at once are shown in red. The interaction network has 64 nodes and 96 edges.
Figure 8. Protein–chemical interaction network. Of a total of 70 nodes, genes are colored in yellow, chemical agents are colored in blue, chemical agents that target more than 2 genes at once are shown in green, and chemical agents that target 5 genes at once are shown in red. The interaction network has 64 nodes and 96 edges. 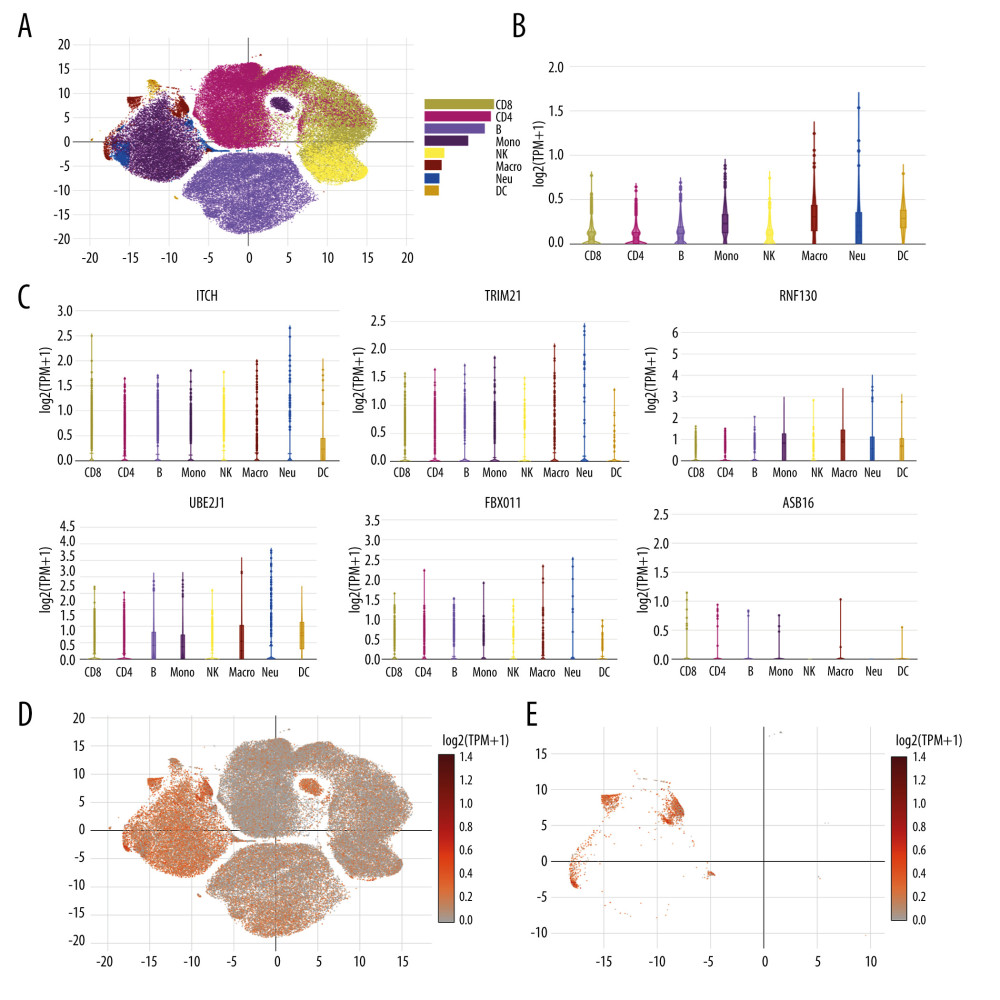 Figure 9. Single cell RNA-seq analysis of COVID-19 patients from online database platform. (A) Eight-cell types were identified by the cell markers; cells were clustered by the tSNE method. (B) Violin plot of the integration of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells. (C) Violin plot of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells, respectively. (D) Scatter plots of all the cells with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression. (E) Scatter plots of macrophages with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression.
Figure 9. Single cell RNA-seq analysis of COVID-19 patients from online database platform. (A) Eight-cell types were identified by the cell markers; cells were clustered by the tSNE method. (B) Violin plot of the integration of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells. (C) Violin plot of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells, respectively. (D) Scatter plots of all the cells with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression. (E) Scatter plots of macrophages with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression. Tables
Table 1. Clinical characteristics of 4 patients with severe COVID-19 and 4 healthy controls.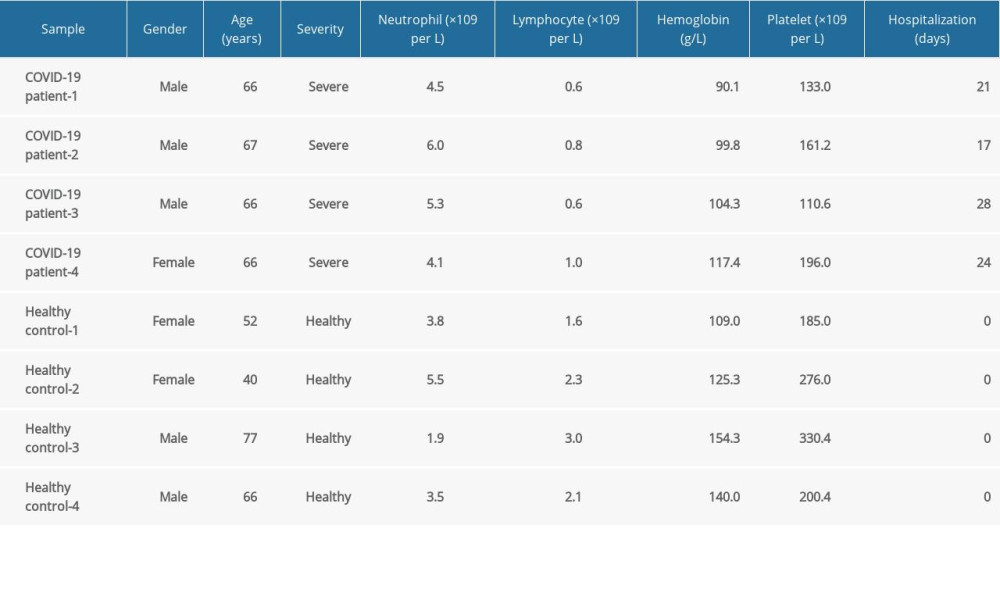 Table 2. The molecular complex detection (MCODE) plug-in of Cytoscape was used to process the data downloaded from STRING; specific gene cluster data were exported and presented in a tabular form.
Table 2. The molecular complex detection (MCODE) plug-in of Cytoscape was used to process the data downloaded from STRING; specific gene cluster data were exported and presented in a tabular form.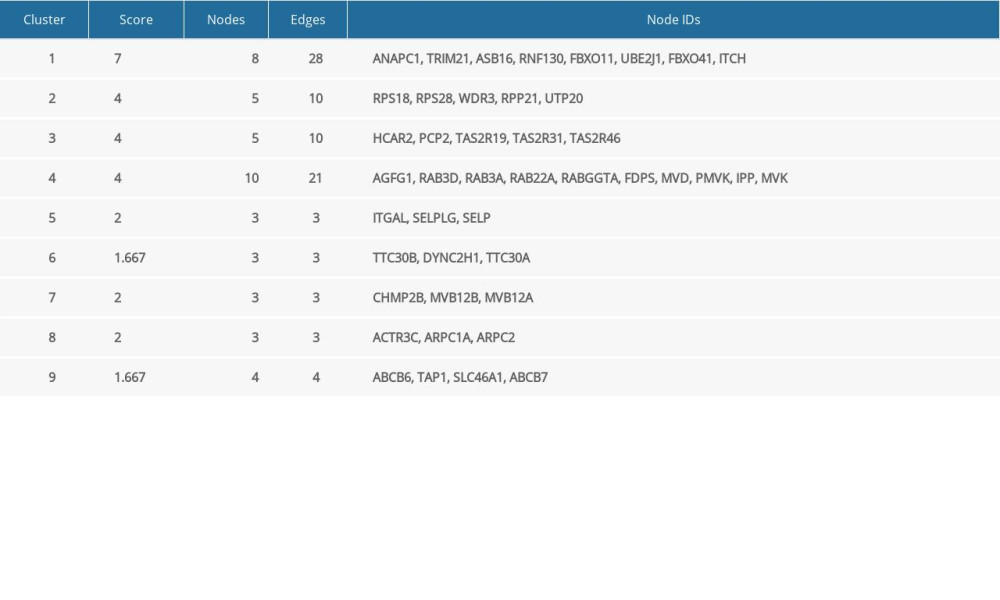 Table 3. Biological processes enrichment analysis of genes in cluster 1.
Table 3. Biological processes enrichment analysis of genes in cluster 1.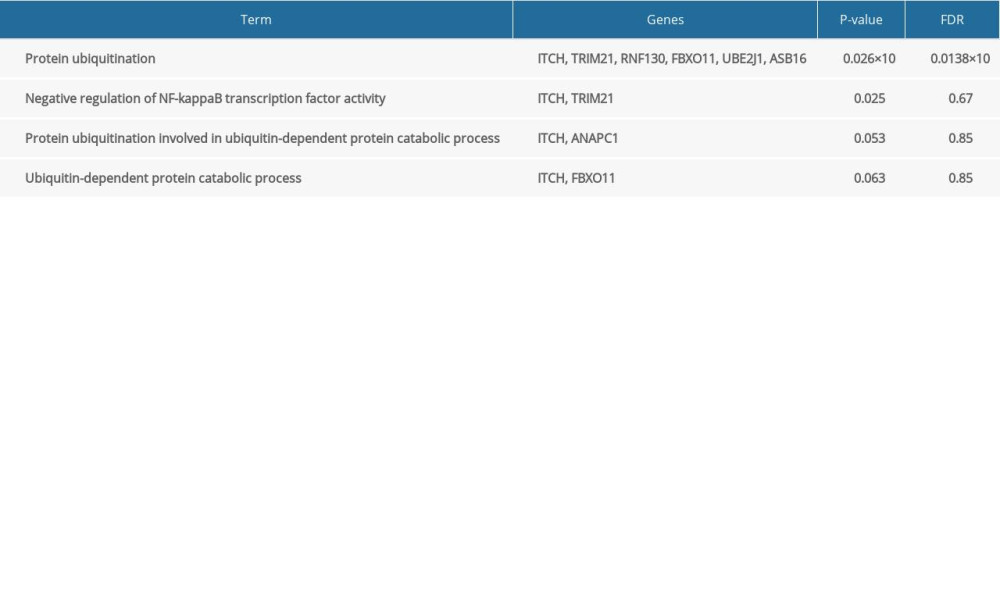
References
1. Bar-On YM, Flamholz A, Phillips R, SARS-CoV-2 (COVID-19) by the numbers: eLife, 2020; 9; e57309
2. Wang D, Hu B, Hu C, Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China: JAMA, 2020; 323(11); 1061-69
3. Wu JT, Leung K, Bushman M, Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China: Nat Med, 2020; 26(4); 506-10
4. Verity R, Okell LC, Dorigatti I, Estimates of the severity of coronavirus disease 2019: A model-based analysis: Lancet Infect Dis, 2020; 20(6); 669-77
5. Lopez Bernal J, Andrews N, Gower C, Effectiveness of COVID-19 vaccines against the B.1.617.2 (delta) variant: N Engl J Med, 2021; 385(7); 585-94
6. Aslam S, Goldstein DR, Vos R, COVID-19 vaccination in our transplant recipients: The time is now: J Heart Lung Transplant, 2021; 40(3); 169-71
7. Nain Z, Rana HK, Liò P, Pathogenetic profiling of COVID-19 and SARS-like viruses: Brief Bioinform, 2021; 22(2); 1175-96
8. Ma M-L, Shi D-W, Li Y, Systematic profiling of SARS-CoV-2-specific IgG responses elicited by an inactivated virus vaccine identifies peptides and proteins for predicting vaccination efficacy: Cell Discov, 2021; 7(1); 67
9. Rabi FA, Al Zoubi MS, Kasasbeh GA, SARS-CoV-2 and coronavirus disease 2019: What we know so far: Pathogens, 2020; 9(3); 231
10. Walsh CT, Garneau-Tsodikova S, Gatto GJ, Protein posttranslational modifications: The chemistry of proteome diversifications: Angew Chem Int Ed Engl, 2005; 44(45); 7342-72
11. Kliza K, Husnjak K, Resolving the complexity of ubiquitin networks: Front Mol Biosci, 2020; 7; 21
12. Rajsbaum R, García-Sastre A, Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways: Trends Microbiol, 2013; 21(8); 421-29
13. Swatek KN, Komander D, Ubiquitin modifications: Cell Res, 2016; 26(4); 399-422
14. Su WC, Yu WY, Huang SH, Ubiquitination of the cytoplasmic domain of influenza a virus M2 protein is crucial for production of infectious virus particles: J Virol, 2018; 92(4); e01972-17
15. Gassen NC, Niemeyer D, Muth D, SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection: Nat Commun, 2019; 10(1); 5770
16. Vadász I, Weiss CH, Sznajder JI, Ubiquitination and proteolysis in acute lung injury: Chest, 2012; 141(3); 763-71
17. Siu K-L, Yuen K-S, Castano-Rodriguez C, Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC: FASEB J, 2019; 33(8); 8865-77
18. Cao Z, Xia H, Rajsbaum R, Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response: Cell Mol Immunol, 2021; 18(3); 746-48
19. Gao M, Labuda T, Xia Y, Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch: Science, 2004; 306(5694); 271-75
20. Heissmeyer V, Macián F, Im SH, Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins: Nat Immunol, 2004; 5(3); 255-65
21. You F, Sun H, Zhou X, PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4: Nat Immunol, 2009; 10(12); 1300-8
22. Oke V, Wahren-Herlenius M, The immunobiology of Ro52 (TRIM21) in autoimmunity: A critical review: J Autoimmun, 2012; 39(1); 77-82
23. Das A, Dinh PX, Pattnaik AK, Trim21 regulates Nmi-IFI35 complex-mediated inhibition of innate antiviral response: Virology, 2015; 485; 383-92
24. Watanabe T, Yamashita S, Ureshino H, Targeting aberrant DNA hypermethylation as a driver of ATL leukemogenesis by using the new oral demethylating agent OR-2100: Blood, 2020; 136(7); 871-84
25. Schneider C, Kon N, Amadori L, FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease: Blood, 2016; 128(5); 660-66
26. Feng T, Deng L, Lu X, Ubiquitin-conjugating enzyme UBE2J1 negatively modulates interferon pathway and promotes RNA virus infection: Virol J, 2018; 15(1); 132
27. Fu T, Ji K, Jin L, ASB16-AS1 up-regulated and phosphorylated TRIM37 to activate NF-κB pathway and promote proliferation, stemness, and cisplatin resistance of gastric cancer: Gastric Cancer, 2021; 24(1); 45-59
28. Yao X, You G, Zhou C, LncRNA ASB16-AS1 promotes growth and invasion of hepatocellular carcinoma through regulating miR-1827/FZD4 axis and activating Wnt/β-catenin pathway: Cancer Manag Res, 2019; 11; 9371-78
29. Zhang D, Zhou H, Liu J, Long noncoding RNA ASB16-AS1 promotes proliferation, migration, and invasion in glioma cells: Biomed Res Int, 2019; 2019; 5437531
30. Raha S, Mallick R, Basak S, Is copper beneficial for COVID-19 patients?: Med Hypotheses, 2020; 142; 109814
31. Sagripanti JL, Routson LB, Lytle CD, Virus inactivation by copper or iron ions alone and in the presence of peroxide: Appl Environ Microbiol, 1993; 59(12); 4374-76
32. Noyce JO, Michels H, Keevil CW, Inactivation of influenza A virus on copper versus stainless steel surfaces: Appl Environ Microbiol, 2007; 73(8); 2748-50
33. van Doremalen N, Bushmaker T, Morris DH, Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1: N Engl J Med, 2020; 382(16); 1564-67
34. Huang J, Wang Y, Zha Y, Transcriptome analysis reveals hub genes regulating autophagy in patients with severe COVID-19: Front Genet, 2022; 13; 908826
35. Zhou Z, Zhou X, Cheng L, Machine learning algorithms utilizing blood parameters enable early detection of immunethrombotic dysregulation in COVID-19: Clin Transl Med, 2021; 11(9); e523
36. Schulte-Schrepping J, Reusch N, Paclik D, Severe COVID-19 is marked by a dysregulated myeloid cell compartment: Cell, 2020; 182(6); 1419-40
37. Guan WJ, Liang WH, Zhao Y, Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis: Eur Respir J, 2020; 55(5); 2000547
38. Fang L, Karakiulakis G, Roth M, Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection?: Lancet Respir Med, 2020; 8(4); e21
39. Mousquer GT, Peres A, Fiegenbaum M, Pathology of TB/COVID-19 co-infection: The phantom menace: Tuberculosis (Edinb), 2021; 126; 102020
40. Zhang H, Zheng H, Zhu J, Ubiquitin-modified proteome of SARS-CoV-2-infected host cells reveals insights into virus–host interaction and pathogenesis: J Proteome Res, 2021; 20(5); 2224-39
41. Gao M, Karin M, Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli: Mol Cell, 2005; 19(5); 581-93
42. Aleebrahim-Dehkordi E, Molavi B, Mokhtari M, T helper type (Th1/Th2) responses to SARS-CoV-2 and influenza A (H1N1) virus: From cytokines produced to immune responses: Transplant Immunol, 2022; 70; 101495
43. Foss S, Bottermann M, Jonsson A, TRIM21 – from intracellular immunity to therapy: Front Immunol, 2019; 10; 2049
44. Stukalov A, Girault V, Grass V, Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV: bioRxiv, 2021 2020.06.17.156455
45. Kaneko N, Kuo HH, Boucau J, Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19: Cell, 2020; 183(1); 143-57e13
46. Kim Y-M, Shin E-C, Type I and III interferon responses in SARS-CoV-2 infection: Exp Mol Med, 2021; 53(5); 750-60
47. Kircheis R, Haasbach E, Lueftenegger D, NF-κB pathway as a potential target for treatment of critical stage COVID-19 patients: Front Immunol, 2020; 11; 598444
48. Islam T, Rahman MR, Aydin B, Integrative transcriptomics analysis of lung epithelial cells and identification of repurposable drug candidates for COVID-19: Eur J Pharmacol, 2020; 887; 173594
49. Lee Yu K, Jung YM, Park SH, Human transcription factor YY1 could upregulate the HIV-1 gene expression: BMB Rep, 2020; 53(5); 248-53
50. Liu H, Xu J, Yang Y, Oncogenic HPV promotes the expression of the long noncoding RNA lnc-FANCI-2 through E7 and YY1: Proc Natl Acad Sci USA, 2021; 118(3); e2014195118
51. Griese M, Zarbock R, Costabel U, GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders: BMC Pulm Med, 2015; 15; 87
52. Wang X, Wen Y, Xie X, Dysregulated hematopoiesis in bone marrow marks severe COVID-19: Cell Discov, 2021; 7(1); 60
53. Wang J, Huang R, Xu Q, Mesenchymal stem cell-derived extracellular vesicles alleviate acute lung injury via transfer of miR-27a-3p: Crit Care Med, 2020; 48(7); e599-e610
54. Cui H, Banerjee S, Xie N, MicroRNA-27a-3p is a negative regulator of lung fibrosis by targeting myofibroblast differentiation: Am J Resp Cell Mol Biol, 2016; 54(6); 843-52
55. Li X, Ma C, Luo H, Identification of the differential expression of genes and upstream microRNAs in small cell lung cancer compared with normal lung based on bioinformatics analysis: Medicine, 2020; 99(11); e19086
56. Qiu X, Lin J, Liang B, Identification of hub genes and microRNAs associated with idiopathic pulmonary arterial hypertension by integrated bioinformatics analyses: Front Genet, 2021; 12; 667406
57. Xu J, Meng Q, Li X, Long noncoding RNA MIR17HG promotes colorectal cancer progression via miR-17-5p: Cancer Res, 2019; 79(19); 4882-95
58. Shi XY, Wang H, Wang W, MiR-98-5p regulates proliferation and metastasis of MCF-7 breast cancer cells by targeting Gab2: Eur Rev Med Pharmacol Sci, 2020; 24(21); 10914
59. Iacona JR, Lutz CS, miR-146a-5p: Expression, regulation, and functions in cancer: Wiley Interdiscip Rev RNA, 2019; 10(4); e1533
60. Zheng Y, Liu J, Chen P, Exosomal miR-22-3p from human umbilical cord blood-derived mesenchymal stem cells protects against lipopolysaccharid-induced acute lung injury: Life Sci, 2021; 269; 119004
61. Zhou Z, Zhu Y, Gao G, Long noncoding RNA SNHG16 targets miR-146a-5p/CCL5 to regulate LPS-induced WI-38 cell apoptosis and inflammation in acute pneumonia: Life Sci, 2019; 228; 189-97
62. Li C, Hu X, Li L, Differential microRNA expression in the peripheral blood from human patients with COVID-19: J Clin Lab Anal, 2020; 34(10); e23590
63. Dyall J, Gross R, Kindrachuk J, Middle East Respiratory Syndrome and Severe Acute Respiratory Syndrome: Current therapeutic options and potential targets for novel therapies: Drugs, 2017; 77(18); 1935-66
64. Li G, De Clercq E, Therapeutic options for the 2019 novel coronavirus (2019-nCoV): Nat Rev Drug Discov, 2020; 19(3); 149-50
65. Guo H, Jian Z, Liu H, TGF-β1-induced EMT activation via both Smad-dependent and MAPK signaling pathways in Cu-induced pulmonary fibrosis: Toxicol Appl Pharmacol, 2021; 418; 115500
66. Lin X, Zhang H, Boyce B, Ubiquitination of interleukin-1α is associated with increased pro-inflammatory polarization of murine macrophages deficient in the E3 ligase ITCH: J Biol Chem, 2020; 295(33); 11764-75
67. Overmyer KA, Shishkova E, Miller IJ, Large-scale multi-omic analysis of COVID-19 severity: Cell Syst, 2021; 12(1); 23-40e7
68. Li CX, Chen J, Lv SK, Whole-transcriptome RNA sequencing reveals significant differentially expressed mRNAs, miRNAs, and lncRNAs and related regulating biological pathways in the peripheral blood of COVID-19 patients: Mediators Inflamm, 2021; 2021; 6635925
Figures
Figure 1. Diagrammatic representation of the overall general process flow of this study showing the RNA sequencing (RNA-Seq) datasets of 4 patients with severe COVID-19 and 4 healthy controls, from whom peripheral blood samples were obtained from GSE167930. Then, the raw data were processed to identify the differentially expressed gene (DEGs). We used these DEGs for Gene Ontology and pathway analysis and protein–protein interaction (PPI) analysis. The most significant gene cluster in the PPI network was identified and used for biological process enrichment analysis. Genes in this cluster were found to play a role in protein ubiquitination. Then, the analysis of genes involved in protein ubiquitination was performed to identify the network of protein-chemical interactions for potent chemical agents, as well as transcription factors and microRNAs at regulatory checkpoints. Ultimately, single-cell RNA-Seq analysis of an online dataset was carried out to reveal the expression of the genes regulating protein ubiquitination that is specific to different cell types.Figure 2. Differentially expressed genes in patients with severe COVID-19 and healthy control samples are shown in the (A) volcano plot and (B) heatmap. H – healthy control; S – severe COVID-19.Figure 3. Gene Ontology and KEGG enrichment analysis of 268 differentially expressed genes (DEGs) in severe COVID-19. The top 10 terms in (A) molecular function, (B) cellular component, and (C) biological process category are presented. The total of 8 significant pathways involved in (D) KEGG enrichment are shown using a scatter plot. Colors represent the P values and sizes of the spots represent the counts of genes.Figure 4. The top 3 pathways in the Reactome database. The gene set was filtered using gene set enrichment analysis, based on the number of genes contained in the gene set, with a minimum number of 15 genes and a maximum number of 500 genes by default. (A) REACTOME_INSULIN_RECEPTOR_RECYCLING, (B) REACTOME_LATENT_INFECTION_OF_HOMO_SAPIENS_WITH MYCOBACTERIUM_TUBERCULOSIS, (C) REACTOME_TRANSFERRIN_ENDOCYTOSIS _AND_RECYCLING.Figure 5. A network of protein–protein interactions (PPI) was built using STRING and Cytoscape. Different colors are used to denote the various clusters that MCODE examined. The PPI network has 261 nodes and 307 edges.Figure 6. The regulatory interaction network of gene-transcription factors was established with network analyst. Genes are represented by circle nodes and are colored orange; transcription factors are represented by square nodes and are colored green; transcription factors that simultaneously target more than 2 genes are represented by blue nodes.Figure 7. Interaction network between genes involved in protein ubiquitination and targeted miRNAs. Genes are shown in blue; miRNAs are shown in pink; miRNAs targeting more than 2 genes simultaneously are shown in green; miRNAs targeting 5 genes simultaneously are shown in yellow. There are 134 nodes and 187 edges.Figure 8. Protein–chemical interaction network. Of a total of 70 nodes, genes are colored in yellow, chemical agents are colored in blue, chemical agents that target more than 2 genes at once are shown in green, and chemical agents that target 5 genes at once are shown in red. The interaction network has 64 nodes and 96 edges.Figure 9. Single cell RNA-seq analysis of COVID-19 patients from online database platform. (A) Eight-cell types were identified by the cell markers; cells were clustered by the tSNE method. (B) Violin plot of the integration of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells. (C) Violin plot of ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression across different cells, respectively. (D) Scatter plots of all the cells with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression. (E) Scatter plots of macrophages with ITCH, TRIM21, RNF130, UBE2J1, FBXO11, and ASB16 expression. Tables
Table 1. Clinical characteristics of 4 patients with severe COVID-19 and 4 healthy controls.Table 2. The molecular complex detection (MCODE) plug-in of Cytoscape was used to process the data downloaded from STRING; specific gene cluster data were exported and presented in a tabular form.Table 3. Biological processes enrichment analysis of genes in cluster 1.Table 1. Clinical characteristics of 4 patients with severe COVID-19 and 4 healthy controls.Table 2. The molecular complex detection (MCODE) plug-in of Cytoscape was used to process the data downloaded from STRING; specific gene cluster data were exported and presented in a tabular form.Table 3. Biological processes enrichment analysis of genes in cluster 1. In Press
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
08 Mar 2024 : Laboratory Research
Evaluation of Retentive Strength of 50 Endodontically-Treated Single-Rooted Mandibular Second Premolars Res...Med Sci Monit In Press; DOI: 10.12659/MSM.944110
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952